
摘要
近年来,环状RNA (circRNAs)被发现通过多种机制,特别是内源性竞争性RNA (ceRNA)机制,在肝细胞癌(HCC)中发挥重要的调节作用。因此,探索肝癌中的circrna具有重要意义。在本研究中,我们利用Cytoscape构建了ceRNA和存活网络。我们还利用R、Perl软件以及Gene Ontology (GO)、Kyoto Encyclopedia of Genes and Genomes (KEGG)等多个在线数据库和平台,对这些基因进行了总体存活、免疫细胞浸润、免疫检查点、通路活性、抗癌药物敏感性分析。最后进行受试者操作特征曲线(receiver operator characteristic curve, ROC)分析,确定基因的诊断价值。KEGG分析显示T细胞受体信号通路是主要富集途径。共筛选出29个与生存和预后相关的基因。结果提示ZNF544、WDR76、ACTG1、RASSF3、E2F3、ASRGL1和POGK与多水平免疫细胞浸润有关。此外,免疫检查点分析筛选出ACTG1、E2F3、RASSF3和WDR76。研究还发现,WDR76、E2F3、ASRGL1和POGK主要激活细胞周期和DNA损伤反应(DDR)途径。结果提示,对曲美替尼、瑞法美替尼(RDEA119)和塞鲁美替尼的敏感性与WDR76的表达相关。ROC分析显示,调控轴上所有基因的曲线下面积(area under the curve, AUC)均大于0.7。确定的hsa_circ_0000417/hsa_circ_0002688/hsa_circ_0001387—hsa-miR-199a-5p—WDR76调控轴可能为HCC的进展、临床诊断和治疗提供新的见解。
类似的内容被其他人浏览


介绍
HCC是全球癌症相关死亡的第三大原因,发病率和死亡率高,特别是在亚洲1,2,3。它是隐匿的,容易转移,由于缺乏早期症状,经常在晚期发现。虽然目前肝癌的治疗方法,如手术切除、肝移植、靶向治疗、免疫治疗、局部治疗等技术已经相对成熟,但这些治疗方法对于晚期肝癌患者往往无效。这些治疗方法也无法延长这些患者的生存时间。因此,寻找新的、更有效的治疗靶点势在必行。非编码rna (ncRNAs)时代的到来为这一领域书写了新的篇章,circRNAs就是ncRNAs的新兴成员之一5,6。
环状rna最初是在一种植物病毒中发现的。然而,由于当时检测技术的限制,circrna主要被认为是传统线性剪接的副产品,其潜力未被发掘。多亏了测序技术,它们的潜力被发现了。环状RNA广泛存在于真核细胞中,具有没有5 '和3 '端的闭环结构,与线性RNA相比,具有对RNA酶的抗性和稳定性。环状rna由反向剪接前体mrna产生,由外显子、内含子或两者组成9,10。环状rna有几种形式,包括外显子环状rna (ecrna)、内含子环状rna (ciRNAs)和外显子内含子环状rna (eiciRNAs)。CiRNAs和eiciRNAs主要调控细胞核内的转录和剪接。此外,ecRNAs被转运出去执行特定的生物学功能,包括作为miRNA和蛋白质海绵或将核苷酸序列翻译成肽和蛋白质11,12,13。circrna也与多种疾病相关,包括心血管疾病、免疫系统疾病和肿瘤14,15,16。
已有证据表明,circRNAs和ceRNA机制参与了包括肝癌起始、肝癌干细胞的干性维持和肝癌进展在内的生物学过程。研究结果建议使用circRNA-miRNA-mRNA调控网络作为治疗肝细胞癌的生物标志物和治疗靶点17,18。近年来,生物信息学分析在科学研究领域越来越受欢迎。因此,本研究采用综合生物信息学分析方法。根据ceRNA机制,通过分析差异表达基因,构建了ceRNA网络。我们还构建了生存网络,分析了生存网络中上调mrna的免疫细胞浸润水平、免疫检查点和通路活性。本研究结果将为HCC的发病机制提供新的视角,提高HCC的诊断和治疗水平。图1给出了本研究的流程图。

本研究流程图。
结果
下载数据及差异基因表达分析
根据确定的筛选条件,我们从Gene Expression Omnibus (GEO)数据库中下载了3个微阵列数据(GSE155949、GSE108724和GSE101728)和1个肝癌RNA-seq数据(TCGA)数据库中下载。数据集的细节如表1所示。差异基因表达分析揭示了36个circRNAs (DEcircRNAs)(图2A)、26个miRNAs (DEmiRNAs)(图2B)和2215个mrna (demmrnas)(图2C)的差异表达。这36个circrna被用于构建ceRNA网络。

差异表达基因的鉴定。(A) DEcircRNAs热图,(B) DEmiRNAs热图,(C) demrna热图。
circrna和mirna的靶标预测
在线数据库共预测了2495个circRNAs靶基因,以及与DEmiRNAs交叉后的6个候选mirna(图3A),分别是hsa-miR-375、hsa-miR-3188、hsa-miR-4270、hsa-miR-221-3p、hsa-miR-532-3p和hsa-miR-199a-5p。此外,数据库预测了6个候选mirna的2470个靶基因,与demrna重叠后鉴定出262个候选mrna(图3B)。

miRNA (A)和mRNA (B)的重叠基因。
构建ceRNA网络
按照ceRNA机制,Perl筛选候选基因,获得构建ceRNA网络所需的输入文件,得到6个circrna、3个mirna和94个mrna。将输入的文件导入Cytoscape软件,构建包含103个节点和104条边的ceRNA网络(图4)。网格中节点的颜色表示基因表达,红色和蓝色分别表示高表达和低表达。不同的节点形状表示不同的基因类型:矩形代表circRNAs,三角形代表miRNAs,圆圈代表mrna。不同颜色的线表示节点之间的连接:紫色粗线表示circRNAs与miRNAs的关系,灰色细线表示miRNAs与mrna的关系。

ceRNA网络。
GO和KEGG富集分析
对ceRNA网络中mrna的GO富集分析得出了28个GO条目。分析揭示了主要富集的生物过程,包括调节蛋白丝氨酸/苏氨酸激酶活性,细胞-基质粘附等(图5A,B)。KEGG富集分析表明,T细胞受体信号通路是主要富集途径(图5C,D)。

ceRNA网络mrna的功能富集分析。(A,B) GO富集分析。(C,D) KEGG富集分析。
生存分析与生存网络构建
对ceRNA网络中的mrna进行生存分析,发现29个生存相关基因(图6)。使用Perl对这29个生存相关基因的上游基因进行匹配,确定它们之间的相互关系,获得构建生存网络所需的输入文件。使用Cytoscape创建了包含61个调控轴的存活网络(图7A)。存活网络中circRNAs高表达基因如表2所示。从存活网络中筛选出8个高表达mrna,分别为ZNF544、WDR76、ACTG1、RASSF3、E2F3、ASRGL1、SUCO和POGK进行后续分析。

ceRNA网络中mrna的总体生存分析。

生存网络构建及通路活性分析。(A)生存网络的构建。(B)存活网络中上调mrna的通路活性分析。
通路活性分析
通路活性分析表明,WDR76、E2F3、ASRGL1和POGK激活了细胞周期和DDR通路(图7B)。WDR76对细胞周期通路的激活作用最为显著。该基因还参与细胞凋亡和上皮-间质转化(EMT)途径的激活,并抑制RAS/MAPK途径。
免疫细胞浸润水平和免疫检查点分析
选取存活预后网络中高表达的8个mrna进行免疫细胞浸润分析。结果表明,ACTG1(图8A)、ASRGL1(图8B)、E2F3(图8C)、POGK(图8D)、RASSF3(图8E)、WDR76(图8F)、ZNF544(图8G)与肝脏肝细胞癌(LIHC)微环境中B细胞、CD4+ T细胞、CD8+ T细胞、中性粒细胞、巨噬细胞和树突状细胞的浸润水平存在显著相关性。但是,不能从数据库中检索SUCO。此外,免疫检查点分析显示,在LIHC中,ACTG1、E2F3、RASSF3和WDR76与程序性细胞死亡1 (PDCD1,图9A)、分化簇274 (CD274,图9B)和细胞毒性T淋巴细胞相关抗原4 (CTLA4,图9C)呈正相关。

存活网络中上调mrna的免疫细胞浸润水平分析。(a) actg1, (b) asrgl1, (c) e2f3, (d) pogk, (e) rassf3, (f) wdr76, (g) znf544。

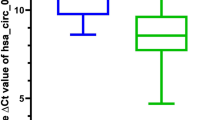
免疫检查点,药物敏感性,ROC分析。ACTG1、E2F3、RASSF3和WDR76分别与PDCD1 (A)、CD274 (B)和CTLA4 (C)的免疫检查点分析。(D)药物敏感性分析。对hsa_circ_0000417 (E)、hsa_circ_0001387 (F)、hsa_circ_0002688 (G)、has-miR-199a-5p (H)和WDR76 (I)进行ROC分析。
药物敏感性及ROC分析
药敏分析显示,WDR76与曲美替尼、RDEA119、塞鲁美替尼的敏感性呈正相关(图9D)。研究结果表明,WDR76可能是存活网络中高表达基因中的枢纽基因。因此,我们从ceRNA网络中确定了hsa_circ_0000417/hsa_circ_0002688/hsa_circ_0001387-hsa_miR-199a-5p-WDR76调控轴。对鉴定出的调控轴上的基因进行ROC分析,以确定其对HCC诊断的价值。auc分别为0.738 (hsa_circ_0000417,图9E)、0.876 (hsa_circ_0001387,图9F)、0.785 (hsa_circ_0002688,图9G)、0.980 (hsa_miR-199a-5p,图9H)和0.844 (WDR76,图9I)。结果表明,这些基因可能用于HCC的诊断标记。并且,我们获得了circRNAs与miRNA之间的潜在结合位点(图10A), hsa_circ_0000417(图10B), hsa_circ_0002688(图10C)和hsa_circ_0001387(图10D)的结构。图10E显示了调节轴的模型。因此,本研究揭示了调节轴与LIHC患者的生存、免疫细胞浸润水平、免疫检查点、通路活性、药物敏感性和HCC诊断之间的联系。同时确定了其他可能参与调节肝细胞癌发生发展并对HCC的诊断和治疗具有参考价值的因素。

circRNA-miRNA的调控轴模型及结合位点。(A) hsa_circ_0000417/0001387/0002688与has-miR-199a-5p之间的结合位点。结构为hsa_circ_0000417(B)/0002688 (C)/0001387 (D), ORF打开读框。(E)调节轴模型。向上的红色箭头表示相应rna的表达上调,向下的箭头表示相反。
讨论
近年来,对circRNAs在肿瘤细胞中的各种机制,特别是ceRNA机制的研究不断扩大。ceRNA机制源于一种假设,即ceRNA可以竞争与靶向mirna结合,减少mirna的数量并增加下游mRNA靶标,从而调节疾病。有大量证据支持ceRNA机制在包括hcc21,22,23在内的多种肿瘤的发生和发展中发挥作用。本研究基于ceRNA机制和生物信息学分析,构建了ceRNA网络。多基因功能分析和鉴定数据库最终确定了HCC的关键调控轴。我们的研究结果将为HCC的发病机制、诊断和治疗提供有价值的参考。
已经确定了调节肿瘤发展的各种生物过程和信号通路。细胞基质粘附在包括浸润性乳腺癌在内的不同肿瘤细胞的侵袭性调节中起着至关重要的作用24,25。此外,T细胞受体信号通路是T细胞部分激活介导免疫应答的细胞内通路。研究发现分化146簇(CD146)联合淋巴细胞激酶(LCK)可促进小鼠T细胞受体信号的启动和抗肿瘤免疫应答26。几乎所有肿瘤的细胞周期通路都是异常的,这可能是肿瘤细胞无限复制的关键原因之一。研究还表明,细胞周期通路的遗传变异可能影响III期和iv期非小细胞肺癌(NSCLC)患者的生存27。细胞周期途径也可能调节前列腺癌的发生,这表明它们有可能作为乳腺癌治疗的靶点。DNA损伤反应是由于各种原因引起的DNA损伤后细胞进行修复的过程。受损的修复过程可能导致突变传播基因组的不稳定,并有利于肿瘤细胞的进展。一项研究发现apurinic apyrimidinic endodeoxyribonuclease 2 (APE2)可以调节DDR通路,维持胰腺癌基因组的完整性30。在胃癌中,DDR可以预测肿瘤进展和临床预后31。这些结果表明,我们研究中发现的ceRNA网络可能促进HCC的发生和HCC细胞的侵袭进展,从而恶化HCC患者的临床预后。生存分析的结果也支持这种临床预后价值。
mirna是一种短的ncrna,可以作为表观遗传调节剂促进或抑制恶性肿瘤(如HCC)的肿瘤发生和进展。在本研究中,我们通过生物信息学分析鉴定了hsa-miR-199a-5p。研究发现,靶向kelch样家族成员3 (KLHL3)的hsa-miR-199a-5p可能参与了NSCLC32的发生发展。发现长链ncRNA (lncRNA) AB209371的高表达有利于肝癌细胞的EMT,也可能是hsa-miR-199a-5p失活的直接原因。沉默AB209371联合过表达hsa-miR-199a-5p可抑制肝癌转移和肝癌细胞的EMT,提示hsa-miR-199a-5p可能作为抑制HCC转移的靶点33。这一发现与我们以前的研究结果一致。免疫细胞浸润是肿瘤微环境的重要组成部分。研究表明,B细胞、CD4+ T细胞、CD8+ T细胞、中性粒细胞、巨噬细胞、树突状细胞等浸润肿瘤的免疫细胞可影响肿瘤患者的免疫治疗效果和预后34,35。Gao等人发现SORT1基因与免疫细胞浸润之间存在关联,提示其有可能作为HCC预测的新生物标志物。干扰SORT1可以显著抑制HCC细胞的生长,提示开发抗癌策略的潜在新靶点,特别是HCC36。一般来说,免疫检查点作为免疫系统的调节器,维持和调节自身免疫和外周组织的免疫过程。然而,肿瘤细胞利用这种机制来逃避免疫37。研究发现多功能适配体(P1/C4-bi-apt)可以阻断CTLA4/B7和PDCD1/CD274信号通路,增强对HCC的免疫应答。该适体在HCC免疫治疗中的潜在应用此前已被探索过38。免疫检查点CD274上调可增加细胞分裂周期相关蛋白2 (CDCA2)的表达,预测HCC39不良预后。我们发现ceRNA网络中的ACTG1、E2F3、RASSF3和WDR76与免疫检查点密切相关,提示ceRNA网络调节HCC细胞的免疫逃逸。同时,药物敏感性分析显示WDR76与曲美替尼、RDEA119、塞鲁美替尼等抗癌药物的敏感性存在相关性。先前的研究表明,利法美替尼联合索拉非尼对失去手术机会的ras突变型HCC患者更有益40。然而,塞鲁美替尼联合索拉非尼治疗HCC的价值与其他单药治疗并无显著差异。已发现曲美替尼在治疗卵巢癌和高、低级别胶质瘤方面有显著效果42,43。然而,这种对HCC的影响尚未见报道。对调节轴上所有基因的ROC分析显示hsa_circ_0000417、hsa_circ_0002688、hsa_circ_0001387、hsa_miR-199a-5p和WDR76对HCC的潜在诊断价值。然而,我们的研究也存在局限性。本研究使用公共数据库、在线平台和其他生物信息学分析来确定特定临床诊断和治疗的价值。本研究缺乏细胞和动物实验来支持研究结果,应纳入未来的研究。
总之,通过分析差异表达的circrna、mirna和mrna,我们成功构建了ceRNA和存活网络。结果显示hsa_circ_0000417/hsa_circ_0002688/hsa_circ_0001387-hsa-miR-199a-5p-WDR76调节轴与HCC患者的生存预后、肿瘤浸润免疫细胞、免疫逃逸、通路活性和药物敏感性相关。研究结果表明,这种调节轴可以作为HCC临床诊断和治疗的新的潜在生物标志物和靶点。这些结果也为HCC的发生和发展机制提供了见解。
材料与方法
数据下载
在GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)中以“肝癌”为关键词搜索相应的circRNA、miRNA和mRNA微阵列数据集。筛选和选择数据集的标准包括包含人类肝癌及其邻近正常肝组织的序列样本。标准组和肿瘤组的样本量均大于6例。据此筛选GSE155949 (circRNAs表达谱芯片)、GSE108724 (miRNAs表达谱芯片)、GSE101728 (mrna表达谱芯片)。筛选探针矩阵和平台文件进行进一步分析。从TCGA数据库(https://portal.gdc.Cancer.gov/)下载HCC表达和临床数据,用于后续的生存分析。
差异表达基因的分析
使用Perl (version 5.32.1001, https://strawberryperl.com/)对下载的芯片数据进行整理,得到相应的基因表达矩阵文件。根据探针矩阵文件划分肿瘤组和正常组。采用R软件(版本4.1.2,https://www.r-project.org/)和Limma软件包对数据进行分析,以调整后的P < 0.05为过滤阈值。最后,测定HCC组和正常组的DEcircRNAs、DEmiRNAs和demmrnas。这些decircrna被用作构建ceRNA网络的候选环状rna。对demirna和demmrna进行进一步分析。
从数据库中预测目标基因
decircrna的靶标使用三个在线数据库进行预测:CircBanK44 (http://www.circbank.cn/)、CircRNA Interactome45 (https://circinteractome.nia.nih.gov/)和Cancer-Specific CircRNA Database46 (http://gb.whu.edu.cn/CSCD/)。用于构建ceRNA网络的候选mirna是通过将三个数据库预测的靶标与demirna交叉获得的。使用miRDB47、48 (http://mirdb.org/)和TargetScanHuman49 (https://www.targetscan.org/vert_72/)数据库进行候选mirna的靶标预测。将预测的靶标与demrna重叠,以获得构建ceRNA网络的候选mrna。
ceRNA网络建设
使用Perl匹配识别的候选基因之间的关系,准备构建ceRNA网络所需的输入文件。候选基因之间的关系应符合ceRNA机制:(1)circRNA表达越高,靶mirna表达越低,下游mrna表达越高;(2) circRNA表达越低,靶mirna表达越高,下游mrna表达越低。使用Cytoscape软件50(版本3.8.0,https://cytoscape.org/)构建ceRNA网络。
GO和KEGG富集分析
我们使用R软件和' org. hs . egg .db '包将ceRNA网络中mrna的基因符号转换为功能富集分析所需的ID。途径富集分析基于Kanehisa实验室开发的KEGG数据库51,52,53。使用“clusterProfiler、ggplot2、org. hh . egg .db、enrichment plot、circlize、RColorBrewer、dplyr、ggpubr和complexHeatmap”软件包进行GO和KEGG富集分析和数据可视化。以P < 0.05为筛选阈值。
生存分析
从TCGA数据库中检索并下载肝细胞癌的转录组和临床数据。用R和Perl软件对数据进行处理,得到基因表达和生存时间文件。使用R软件和“Survival, survminer”软件包对ceRNA网络中的mrna进行生存分析,以筛选与HCC患者生存预后相关的mrna。P < 0.05提示mRNA与HCC患者生存预后有相关性。
构建生存网络
利用从上述步骤中获得的生存相关mrna,获得了它们的上游相关mirna和circRNA的信息。相应准备了网络关系、节点等相关输入文件。细胞景观构建存活网络。使用circprimer2.0软件检索存活网络中circrna的结构、位置和其他信息54。
通路活性分析
基因集癌症分析(GSCALite)55是基于网络和多数据库的癌症基因组分析平台(http://bioinfo.life.hust.edu.cn/web/GSCALite/)。使用该平台分析了生存网络通路中的8个高表达mrna。根据需要输入生存相关基因,选择TCGA表达数据,癌症类型选择LIHC。通过通路活性分析模块进行分析,得出最终结果。
免疫细胞浸润水平和免疫检查点分析
肿瘤免疫估计资源(TIMER)56是一个公共网络平台(https://cistrome.shinyapps.io/timer/),用于分析不同类型癌症的免疫细胞浸润水平。选择存活网络中高表达的8个mrna,分析肝细胞癌免疫细胞浸润水平。这些基因被输入到搜索框中,LIHC被选为癌症类型。选择免疫浸润相关细胞(B细胞、CD4+ T细胞、CD8+ T细胞、中性粒细胞、巨噬细胞和树突状细胞)进行相关性分析。基因表达谱交互分析(GEPIA)57是一个多功能在线平台(http://gepia.cancer-pku.cn/about.html),用于分析肿瘤和正常细胞之间基因的差异表达。该平台还可用于与所选癌症类型相关的生存分析、相似基因检测、相关分析、降维分析等。本研究利用GEPIA的相关分析模块,确定了8种mrna的表达与肿瘤免疫逃逸相关的3个经典免疫检查点PDCD1、CD274和CTLA4的相关性。癌症类型选择LIHC,相关系数采用Pearson。
药物敏感性分析
GSCALite分析平台整合了癌症药物敏感性基因组学(Genomics of drug sensitivity in cancer, GDSC)和癌症治疗反应门户网站(cancer Therapeutics Response Portal, CTRP)中癌细胞系的药物敏感性和基因表达谱数据。我们对8种与生存相关的高表达mrna进行了药物敏感性分析。肿瘤类型选择LIHC,药敏模块选择GDSC,肿瘤药敏数据集选择GDSC,对半数最大抑制浓度(IC50)进行Spearman相关分析。调整参数后,结果生成。
ROC分析
采用R软件和“pROC”软件包进行ROC分析,明确circRNA-miRNA-mRNA调控轴上基因对HCC的诊断价值。HCC组织和对照组的表达数据使用上述分析中调整的三个微阵列数据进行分析,AUC≥0.7被认为具有HCC的诊断价值。所有统计分析均使用R软件或在线平台进行。
结合位点的确定和调控轴的模型构建
利用circprimer2.0软件查询调控轴上3种circrna的相关信息,得到它们的序列。从miRWalk (version 3, http://mirwalk.umm.uni-heidelberg.de/)下载hsa-miR-199a-5p序列,并通过序列比对找到circRNAs与miRNA之间的潜在结合位点。同时,我们使用Microsoft PowerPoint软件(版本2021,https://www.microsoft.com/zh-cn/microsoft-365/powerpoint)绘制了监管轴的模型图,以显示这些元素之间的控制关系。
道德声明
本研究使用的数据均来自公共数据库,没有进行任何与动物或人体标本相关的实验,无需伦理审批。
数据可用性
本文使用的数据集和数据均来自GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155949,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108724,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101728)和TCGA数据库(https://portal.gdc.Cancer.gov/)。
参考文献
西格尔,r.l.,米勒,k.d., Fuchs, h.e.和Jemal, A.癌症统计,2022。中国临床医学杂志,2002,7 - 12。https://doi.org/10.3322/caac.21708(2022)。
远东和东南亚患者肝细胞癌的转移流行病学:解释和意义。咕咕叫。肿瘤防治杂志。众议员24,187-193。https://doi.org/10.1007/s11912 - 021 - 01160 - 5(2022)。
张春华,程艳,张淑娟,范静,高强。亚洲地区肝细胞癌流行病学的变化。中华肝病杂志,42,2029-2041。https://doi.org/10.1111/liv.15251(2022)。
Moon, H.等。肝细胞癌从最初诊断到死亡的各种治疗方法:累积治疗的观察。J.癌症与临床。医学杂志,143,2327-2339。https://doi.org/10.1007/s00432 - 017 - 2480 - 9(2017)。
许明杰,尹杰,金世国,非编码rna在肝脏疾病进展为肝细胞癌中的作用。拱门。调药的。Res. 42,48 - 62。https://doi.org/10.1007/s12272 - 018 - 01104 - x(2019)。
非编码RNA:RNA在癌症中的调控网络。Int。科学通报;https://doi.org/10.3390/ijms19051310(2018)。
Sanger, H. L, Klotz, G., Riesner, D., Gross, H. J. & Kleinschmidt, A. K.类病毒是一种单链共价封闭的环状RNA分子,以高度碱基配对的棒状结构存在。Proc。国家的。学会科学。美国学报73,3852-3856。https://doi.org/10.1073/pnas.73.11.3852(1976)。
荣,D.等。环状rna在肿瘤临床应用中的新见解。Onco。目标,10,2183-2188。https://doi.org/10.2147/ott.S134403(2017)。
Wang, Y.和Wang, Z.有效的反向剪接产生可翻译的环状mrna。RNA(纽约,纽约州)21,172-179。https://doi.org/10.1261/rna.048272.114(2015)。
沃,J. N.等。环状RNA在癌症中的作用。176号牢房,869-881.e813。https://doi.org/10.1016/j.cell.2018.12.021(2019)。
张晓光等。互补序列介导的外显子环状化。159号,134-147号牢房。https://doi.org/10.1016/j.cell.2014.09.001(2014)。
Salzman, J, Gawad, C, Wang, P. L, Lacayo, N.和Brown, P. O.环状rna是不同细胞类型中数百种人类基因的主要转录异构体。科学通报,1(2):393 - 393。https://doi.org/10.1371/journal.pone.0030733(2012)。
Conn, V. M.等。SEPALLATA3中的circRNA通过r环的形成调节其同源mRNA的剪接。植物学报3,1753。https://doi.org/10.1038/nplants.2017.53(2017)。
Lu, D.和Thum, T.基于rna的心血管疾病诊断和治疗策略。Nat. Rev. Cardiol. 16, 661-674。https://doi.org/10.1038/s41569 - 019 - 0218 - x(2019)。
周忠,孙斌,黄松,赵丽。环状rna在免疫调节和自身免疫性疾病中的作用。细胞死亡杂志10,503https://doi.org/10.1038/s41419 - 019 - 1744 - 5(2019)。
孟,S.等。CircRNA:一种新的潜在癌症生物标志物的功能和特性摩尔。癌症16,94。https://doi.org/10.1186/s12943 - 017 - 0663 - 2(2017)。
Li, P.等。circMRPS35促进肝细胞癌的恶性进展和顺铂耐药。摩尔。其他。j。Soc。Gene Ther. 30, 431-447。https://doi.org/10.1016/j.ymthe.2021.08.027(2022)。
Lyu, L.等。血浆外泌体hsa_circ_0070396对肝细胞癌的诊断价值。Biomark。医学,15,359-371。https://doi.org/10.2217/bmm - 2020 - 0476(2021)。
王杰,王春丽,杨丽丽,李凯。基于综合生物信息学分析的肝细胞癌关键基因和mirna鉴定。地中海,杂志。(英国伦敦诺斯伍德)39,21。https://doi.org/10.1007/s12032 - 021 - 01622 - 7(2022)。
Salmena, L., Poliseno, L., Tay, Y., Kats, L.和Pandolfi, P. P.一个ceRNA假说:隐藏RNA语言的罗塞塔石碑?146室,353-358。https://doi.org/10.1016/j.cell.2011.07.014(2011)。
刘婷婷等。肺腺癌cdk2相关免疫预测模型及ceRNA的鉴定——泛癌分析前面。生物工程学报,2002,38(2):481 - 481。https://doi.org/10.3389/fcell.2021.682002(2021)。
Wang, H.等。stat3介导上调lncRNA HOXD-AS1作为ceRNA通过调节SOX4促进肝癌转移。摩尔,癌症16,136。https://doi.org/10.1186/s12943 - 017 - 0680 - 1(2017)。
杨学忠等。LINC01133作为ceRNA通过海绵miR-106a-3p调节APC表达和Wnt/β-catenin通路抑制胃癌进展。摩尔,癌症17,126。https://doi.org/10.1186/s12943 - 018 - 0874 - 1(2018)。
布莱克斯通,b.n.等。肌钙素耗竭可提高乳腺癌细胞的黏附激酶和帕罗西林磷酸化,并增强细胞-基质黏附。点。j .杂志。中国生物医学工程学报,2016,33(4):442 - 449。https://doi.org/10.1152/ajpcell.00276.2014(2015)。
Cagigas, m.l.等。相关的冷冻- et鉴定了在癌细胞中介导细胞-底物粘附和细胞增殖的机械敏感性的肌动蛋白/原肌球蛋白丝。Nat, Mater, 21, 120-128。https://doi.org/10.1038/s41563 - 021 - 01087 - z(2022)。
Duan, H.等。与LCK结合的CD146促进小鼠T细胞受体信号传导和抗肿瘤免疫反应。j .中国。投资。https://doi.org/10.1172/jci148568(2021)。
尹,J.等。细胞周期通路中常见的遗传变异与III-IV期非小细胞肺癌的生存相关。癌变32,1867-1871。https://doi.org/10.1093/carcin/bgr217(2011)。
廖,等。腺苷琥珀酸裂解酶(ADSL)通过细胞周期途径在前列腺癌发生和进展中的致癌作用。癌症杂志,21,467。https://doi.org/10.1186/s12935 - 021 - 02174 - 6(2021)。
孙晓明,王志明,陈晓明,沈凯。CRISPR-cas9筛选乳腺癌细胞周期通路中富集的致死性基因及其预后意义。前面。细胞发育生物学杂志9,646774。https://doi.org/10.3389/fcell.2021.646774(2021)。
侯赛因,m.a.等。在胰腺癌细胞中,APE2是ATR-Chk1 DNA损伤反应通路的一般调节因子,以维持基因组的完整性。前面。细胞发育生物学杂志9,738502。https://doi.org/10.3389/fcell.2021.738502(2021)。
徐晓斌等。DNA损伤反应通路及自噬标志物在胃癌中的表达及预后意义。肿瘤68,1310-1319。https://doi.org/10.4149/neo_2021_210515N667(2021)。
张伟等。利用生物学信息分析非小细胞肺癌患者血浆中潜在的miRNA-mRNA调控网络。癌症22,299。https://doi.org/10.1186/s12885 - 022 - 09281 - 1(2022)。
肖,C.等。LncRNA-AB209371促进肝癌细胞上皮-间质转化。肿瘤防治杂志。众议员41,2957-2966。https://doi.org/10.3892/or.2019.7045(2019)。
爸爸,B. B.等。利用与炎症反应相关的基因标记预测肝癌预后和免疫细胞浸润。第一版。数学。方法医学杂志,2022,2415129。https://doi.org/10.1155/2022/2415129(2022)。
蒋旸,等。肝细胞癌的免疫细胞浸润与免疫治疗。数学。Biosci。工程学报,19,7178-7200。https://doi.org/10.3934/mbe.2022339(2022)。
高旸,等。Sortilin 1通过调节免疫细胞浸润促进肝癌细胞增殖和迁移。[j] .中国生物医学工程学报,2016,35(5):591 - 591。https://doi.org/10.1155/2022/6509028(2022)。
Santos, p.m. & Butterfield, l.h.肝细胞癌免疫检查点的下一步研究。胃肠病学杂志155,1684-1686。https://doi.org/10.1053/j.gastro.2018.11.008(2018)。
Du, Y.等。一种高度稳定的多功能适配体,通过阻断双重免疫检查点增强抗肝癌的抗肿瘤免疫。Biomater。科学9,4159-4168。https://doi.org/10.1039/d0bm02210a(2021)。
唐,M,廖,M, Ai, x &他g .增加CDCA2水平与肝细胞癌预后不良有关,与老年病相关免疫检查点。前面。医学8,773724。https://doi.org/10.3389/fmed.2021.773724(2021)。
林海英等。一项关于MEK抑制剂refametinib (BAY 86-9766)与索拉非尼联合治疗亚洲不可切除肝细胞癌患者疗效和安全性的II期研究。中国。癌症杂志,20,5976-5985。https://doi.org/10.1158/1078 - 0432. - ccr - 13 - 3445(2014)。
Tai, W. M.等。selumetinib (AZD6244, ry -142886)联合索拉非尼治疗晚期肝细胞癌(HCC)的Ib期研究。安。27, 2210-2215。https://doi.org/10.1093/annonc/mdw415(2016)。
Gershenson博士等人。曲美替尼与复发性低级别浆液性卵巢癌患者的标准治疗(GOG 281/LOGS):一项国际、随机、开放标签、多中心、2/3期试验。柳叶刀399,541-553。https://doi.org/10.1016/s0140 - 6736(21) 02175 - 9(2022)。
温,P. Y.等。达非尼联合曲美替尼治疗BRAF(V600E)突变型低级别和高级别胶质瘤(ROAR)患者:一项多中心、开放标签、单臂、2期一揽子试验医学杂志,23,53-64。https://doi.org/10.1016/s1470 - 2045(21) 00578 - 7(2022)。
刘敏,王强,沈健,杨宝彬,丁旭,Circbank:一个具有标准命名法的circRNA综合数据库。中国生物医学工程学报,32(2):444 - 444。https://doi.org/10.1080/15476286.2019.1600395(2019)。
Dudekula, d.b.等人。CircInteractome:一个用于探索环状rna及其相互作用蛋白和微rna的网络工具。RNA生物学报,13,34-42。https://doi.org/10.1080/15476286.2015.1128065(2016)。
夏,S.等。CSCD:癌症特异性环状rna数据库。核酸学报,46,D925-d929。https://doi.org/10.1093/nar/gkx863(2018)。
Chen, Y.和Wang, X. miRDB:一个预测功能性microRNA靶点的在线数据库。核酸学报,48(4):1227 - 1231。https://doi.org/10.1093/nar/gkz757(2020)。
刘伟,王欣。基于microRNA结合和靶标表达数据整合建模的功能性microRNA靶标预测。基因组生物学。20,18。https://doi.org/10.1186/s13059 - 019 - 1629 - z(2019)。
Agarwal, V., Bell, G. W., Nam, J. W. & Bartel, D. P.预测哺乳动物mrna的有效microRNA靶点。Elife https://doi.org/10.7554/eLife.05005(2015)。
香农,P.等。Cytoscape:生物分子相互作用网络集成模型的软件环境。Genome Res. 13, 2498-2504。https://doi.org/10.1101/gr.1239303(2003)。
Kanehisa, M, Furumichi, M, Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG:整合病毒和细胞有机体。核酸学报,49,D545-d551。https://doi.org/10.1093/nar/gkaa970(2021)。
迈向了解细胞有机体的起源与演化。蛋白质科学28,1947-1951。https://doi.org/10.1002/pro.3715(2019)。
Kanehisa, M. & Goto, S. KEGG:京都基因和基因组百科全书。核酸学报,28,27-30。https://doi.org/10.1093/nar/28.1.27(2000)。
钟生和冯杰。CircPrimer 2.0:一个用于circrna注释和预测circrna翻译潜力的软件。中华生物医学杂志。23,215。https://doi.org/10.1186/s12859 y - 022 - 04705 -(2022)。
刘家杰等。GSCALite:用于基因集癌症分析的web服务器。生物信息学学报,34(4):771 - 772。https://doi.org/10.1093/bioinformatics/bty411(2018)。
李,T.等。TIMER:用于肿瘤浸润性免疫细胞综合分析的web服务器。可以。Res. 77, e108-e110。https://doi.org/10.1158/0008 - 5472. - 17 - 0307(2017)。
Tang, Z.等。GEPIA:一个用于癌症和正常基因表达谱分析和交互分析的web服务器。中国生物医学工程学报,32(5):998 - 9102。https://doi.org/10.1093/nar/gkx247(2017)。
致谢
本研究中使用的微阵列分析数据来自GEO数据库:https://www.ncbi.nlm.nih.gov/geo/。与生存分析相关的结果基于TCGA研究网络:https://www.cancer.gov/tcga生成的数据。我们非常感谢提供上述数据的数据库和相关研究团队。
资金
本工作获右江民族医学院研究生创新项目(YXCXJH2022003)资助。
作者信息
作者及单位
贡献
y.l.:下载和整理文献作品和数据集;g.z.:概念化、方法论、软件、可视化和写作;本文的修改和最终批准。
相应的作者
道德声明
相互竞争的利益
作者声明没有利益冲突。
额外的信息
出版商的注意
施普林格·自然对已出版的地图和机构关系中的管辖权要求保持中立。
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议,该协议允许以任何媒介或格式使用、共享、改编、分发和复制,只要您适当地注明原作者和来源,提供知识共享许可协议的链接,并注明是否进行了更改。本文中的图像或其他第三方材料包含在文章的知识共享许可协议中,除非在材料的署名中另有说明。如果材料未包含在文章的知识共享许可中,并且您的预期用途不被法律法规允许或超过允许的用途,您将需要直接获得版权所有者的许可。要查看本许可的副本,请访问http://creativecommons.org/licenses/by/4.0/。
关于本文

引用本文
钟刚,林毅,黄忠。基于生物信息学分析的肝癌circRNA-miRNA-mRNA调控轴的鉴定。科学通报13,37(2023)。https://doi.org/10.1038/s41598-023-30567-2
收稿日期:2022年10月22日
录用日期:2023年2月25日
发布日期:2023年3月6日
DOI: https://doi.org/10.1038/s41598 - 023 - 30567 - 2
这篇文章是由
-
环状RNA circDLG1通过调节miR-141-3p/WTAP轴参与HCC进展
功能与整合基因组学(2023)
评论
通过提交评论,您同意遵守我们的条款和社区准则。如果你发现一些滥用或不符合我们的条款或指导方针,请标记为不适当。
